Project F6620A services include NGS sequencing of the V1V3 region of the 16S rRNA gene amplicons from the samples. First and foremost, please
download this report, as well as the sequence raw data from the download links provided below.
These links will expire after 60 days. We cannot guarantee the availability of your data after 60 days.
Full Bioinformatics analysis service was requested. We provide many analyses, starting from the raw sequence quality and noise filtering, pair reads merging, as well as chimera filtering for the sequences, using the
DADA2 denosing algorithm and pipeline.
We also provide many downstream analyses such as taxonomy assignment, alpha and beta diversity analyses, and differential abundance analysis.
For taxonomy assignment, most informative would be the taxonomy barplots. We provide an interactive barplots to show the relative abundance of microbes at different taxonomy levels (from Phylum to species) that you can choose.
If you specify which groups of samples you want to compare for differential abundance, we provide both ANCOM and LEfSe differential abundance analysis.
The samples were processed and analyzed with the ZymoBIOMICS® Service: Targeted
Metagenomic Sequencing (Zymo Research, Irvine, CA).
DNA Extraction: If DNA extraction was performed, one of three different DNA
extraction kits was used depending on the sample type and sample volume and were
used according to the manufacturer’s instructions, unless otherwise stated. The kit used
in this project is marked below:
☐
ZymoBIOMICS® DNA Miniprep Kit (Zymo Research, Irvine, CA)
☐
ZymoBIOMICS® DNA Microprep Kit (Zymo Research, Irvine, CA)
☐
ZymoBIOMICS®-96 MagBead DNA Kit (Zymo Research, Irvine, CA)
☑
N/A (DNA Extraction Not Performed)
Elution Volume: 50µL
Additional Notes: NA
Targeted Library Preparation: The DNA samples were prepared for targeted
sequencing with the Quick-16S™ NGS Library Prep Kit (Zymo Research, Irvine, CA).
These primers were custom designed by Zymo Research to provide the best coverage
of the 16S gene while maintaining high sensitivity. The primer sets used in this project
are marked below:
☐
Quick-16S™ Primer Set V1-V2 (Zymo Research, Irvine, CA)
☑
Quick-16S™ Primer Set V1-V3 (Zymo Research, Irvine, CA)
☐
Quick-16S™ Primer Set V3-V4 (Zymo Research, Irvine, CA)
☐
Quick-16S™ Primer Set V4 (Zymo Research, Irvine, CA)
☐
Quick-16S™ Primer Set V6-V8 (Zymo Research, Irvine, CA)
☐
Other: NA
Additional Notes: NA
The sequencing library was prepared using an innovative library preparation process in
which PCR reactions were performed in real-time PCR machines to control cycles and
therefore limit PCR chimera formation. The final PCR products were quantified with
qPCR fluorescence readings and pooled together based on equal molarity. The final
pooled library was cleaned up with the Select-a-Size DNA Clean & Concentrator™
(Zymo Research, Irvine, CA), then quantified with TapeStation® (Agilent Technologies,
Santa Clara, CA) and Qubit® (Thermo Fisher Scientific, Waltham, WA).
Control Samples: The ZymoBIOMICS® Microbial Community Standard (Zymo
Research, Irvine, CA) was used as a positive control for each DNA extraction, if
performed. The ZymoBIOMICS® Microbial Community DNA Standard (Zymo Research,
Irvine, CA) was used as a positive control for each targeted library preparation.
Negative controls (i.e. blank extraction control, blank library preparation control) were
included to assess the level of bioburden carried by the wet-lab process.
Sequencing: The final library was sequenced on Illumina® MiSeq™ with a V3 reagent kit
(600 cycles). The sequencing was performed with 10% PhiX spike-in.
The complete report of your project, including all links in this report, can be downloaded by clicking the link provided below. The downloaded file is a compressed ZIP file and once unzipped, open the file “REPORT.html” (may only shown as "REPORT" in your computer) by double clicking it. Your default web browser will open it and you will see the exact content of this report.
Please download and save the file to your computer storage device. The download link will expire after 60 days upon your receiving of this report.
Complete report download link:
To view the report, please follow the following steps:
1.
Download the .zip file from the report link above.
2.
Extract all the contents of the downloaded .zip file to your desktop.
3.
Open the extracted folder and find the "REPORT.html" (may shown as only "REPORT").
4.
Open (double-clicking) the REPORT.html file. Your default browser will open the top age of the complete report. Within the
report, there are links to view all the analyses performed for the project.
The raw NGS sequence data is available for download with the link provided below. The data is a compressed ZIP file and can be unzipped to individual sequence files.
Since this is a pair-end sequencing, each of your samples is represented by two sequence files, one for READ 1,
with the file extension “*_R1.fastq.gz”, another READ 2, with the file extension “*_R1.fastq.gz”.
The files are in FASTQ format and are compressed. FASTQ format is a text-based data format for storing both a biological sequence
and its corresponding quality scores. Most sequence analysis software will be able to open them.
The Sample IDs associated with the R1 and R2 fastq files are listed in the table below:
Sample ID
Original Sample ID
Read 1 File Name
Read 2 File Name
F6620.S100
B3_39
zr6620_100V1V3_R1.fastq.gz
zr6620_100V1V3_R2.fastq.gz
F6620.S101
L1_39
zr6620_101V1V3_R1.fastq.gz
zr6620_101V1V3_R2.fastq.gz
F6620.S102
L2_39
zr6620_102V1V3_R1.fastq.gz
zr6620_102V1V3_R2.fastq.gz
F6620.S103
L3_39
zr6620_103V1V3_R1.fastq.gz
zr6620_103V1V3_R2.fastq.gz
F6620.S104
C1_39
zr6620_104V1V3_R1.fastq.gz
zr6620_104V1V3_R2.fastq.gz
F6620.S105
C2_39
zr6620_105V1V3_R1.fastq.gz
zr6620_105V1V3_R2.fastq.gz
F6620.S106
C3_39
zr6620_106V1V3_R1.fastq.gz
zr6620_106V1V3_R2.fastq.gz
F6620.S107
FB1_46
zr6620_107V1V3_R1.fastq.gz
zr6620_107V1V3_R2.fastq.gz
F6620.S108
FB2_46
zr6620_108V1V3_R1.fastq.gz
zr6620_108V1V3_R2.fastq.gz
F6620.S109
FB3_46
zr6620_109V1V3_R1.fastq.gz
zr6620_109V1V3_R2.fastq.gz
F6620.S010
FA1_4
zr6620_10V1V3_R1.fastq.gz
zr6620_10V1V3_R2.fastq.gz
F6620.S110
FA1_46
zr6620_110V1V3_R1.fastq.gz
zr6620_110V1V3_R2.fastq.gz
F6620.S111
FA2_46
zr6620_111V1V3_R1.fastq.gz
zr6620_111V1V3_R2.fastq.gz
F6620.S112
FA3_46
zr6620_112V1V3_R1.fastq.gz
zr6620_112V1V3_R2.fastq.gz
F6620.S113
B1_46
zr6620_113V1V3_R1.fastq.gz
zr6620_113V1V3_R2.fastq.gz
F6620.S114
B2_46
zr6620_114V1V3_R1.fastq.gz
zr6620_114V1V3_R2.fastq.gz
F6620.S115
B3_46
zr6620_115V1V3_R1.fastq.gz
zr6620_115V1V3_R2.fastq.gz
F6620.S116
L1_46
zr6620_116V1V3_R1.fastq.gz
zr6620_116V1V3_R2.fastq.gz
F6620.S117
L2_46
zr6620_117V1V3_R1.fastq.gz
zr6620_117V1V3_R2.fastq.gz
F6620.S118
L3_46
zr6620_118V1V3_R1.fastq.gz
zr6620_118V1V3_R2.fastq.gz
F6620.S119
C1_46
zr6620_119V1V3_R1.fastq.gz
zr6620_119V1V3_R2.fastq.gz
F6620.S011
FA2_4
zr6620_11V1V3_R1.fastq.gz
zr6620_11V1V3_R2.fastq.gz
F6620.S120
C2_46
zr6620_120V1V3_R1.fastq.gz
zr6620_120V1V3_R2.fastq.gz
F6620.S121
C3_46
zr6620_121V1V3_R1.fastq.gz
zr6620_121V1V3_R2.fastq.gz
F6620.S122
FB1_53
zr6620_122V1V3_R1.fastq.gz
zr6620_122V1V3_R2.fastq.gz
F6620.S123
FB2_53
zr6620_123V1V3_R1.fastq.gz
zr6620_123V1V3_R2.fastq.gz
F6620.S124
FB3_53
zr6620_124V1V3_R1.fastq.gz
zr6620_124V1V3_R2.fastq.gz
F6620.S125
FA1_53
zr6620_125V1V3_R1.fastq.gz
zr6620_125V1V3_R2.fastq.gz
F6620.S126
FA2_53
zr6620_126V1V3_R1.fastq.gz
zr6620_126V1V3_R2.fastq.gz
F6620.S127
FA3_53
zr6620_127V1V3_R1.fastq.gz
zr6620_127V1V3_R2.fastq.gz
F6620.S128
B1_53
zr6620_128V1V3_R1.fastq.gz
zr6620_128V1V3_R2.fastq.gz
F6620.S129
B2_53
zr6620_129V1V3_R1.fastq.gz
zr6620_129V1V3_R2.fastq.gz
F6620.S012
FA3_4
zr6620_12V1V3_R1.fastq.gz
zr6620_12V1V3_R2.fastq.gz
F6620.S130
B3_53
zr6620_130V1V3_R1.fastq.gz
zr6620_130V1V3_R2.fastq.gz
F6620.S131
L1_53
zr6620_131V1V3_R1.fastq.gz
zr6620_131V1V3_R2.fastq.gz
F6620.S132
L2_53
zr6620_132V1V3_R1.fastq.gz
zr6620_132V1V3_R2.fastq.gz
F6620.S133
L3_53
zr6620_133V1V3_R1.fastq.gz
zr6620_133V1V3_R2.fastq.gz
F6620.S134
C1_53
zr6620_134V1V3_R1.fastq.gz
zr6620_134V1V3_R2.fastq.gz
F6620.S135
C2_53
zr6620_135V1V3_R1.fastq.gz
zr6620_135V1V3_R2.fastq.gz
F6620.S136
C3_53
zr6620_136V1V3_R1.fastq.gz
zr6620_136V1V3_R2.fastq.gz
F6620.S137
CF1_53
zr6620_137V1V3_R1.fastq.gz
zr6620_137V1V3_R2.fastq.gz
F6620.S138
CF2_53
zr6620_138V1V3_R1.fastq.gz
zr6620_138V1V3_R2.fastq.gz
F6620.S139
CF3_53
zr6620_139V1V3_R1.fastq.gz
zr6620_139V1V3_R2.fastq.gz
F6620.S013
B1_4
zr6620_13V1V3_R1.fastq.gz
zr6620_13V1V3_R2.fastq.gz
F6620.S014
B2_4
zr6620_14V1V3_R1.fastq.gz
zr6620_14V1V3_R2.fastq.gz
F6620.S015
B3_4
zr6620_15V1V3_R1.fastq.gz
zr6620_15V1V3_R2.fastq.gz
F6620.S016
L1_4
zr6620_16V1V3_R1.fastq.gz
zr6620_16V1V3_R2.fastq.gz
F6620.S017
L2_4
zr6620_17V1V3_R1.fastq.gz
zr6620_17V1V3_R2.fastq.gz
F6620.S018
L3_4
zr6620_18V1V3_R1.fastq.gz
zr6620_18V1V3_R2.fastq.gz
F6620.S019
C1_4
zr6620_19V1V3_R1.fastq.gz
zr6620_19V1V3_R2.fastq.gz
F6620.S001
SupraC1
zr6620_1V1V3_R1.fastq.gz
zr6620_1V1V3_R2.fastq.gz
F6620.S020
C2_4
zr6620_20V1V3_R1.fastq.gz
zr6620_20V1V3_R2.fastq.gz
F6620.S021
C3_4
zr6620_21V1V3_R1.fastq.gz
zr6620_21V1V3_R2.fastq.gz
F6620.S022
CC1_4
zr6620_22V1V3_R1.fastq.gz
zr6620_22V1V3_R2.fastq.gz
F6620.S023
CC2_4
zr6620_23V1V3_R1.fastq.gz
zr6620_23V1V3_R2.fastq.gz
F6620.S024
CC3_4
zr6620_24V1V3_R1.fastq.gz
zr6620_24V1V3_R2.fastq.gz
F6620.S025
CC4_4
zr6620_25V1V3_R1.fastq.gz
zr6620_25V1V3_R2.fastq.gz
F6620.S026
CC5_4
zr6620_26V1V3_R1.fastq.gz
zr6620_26V1V3_R2.fastq.gz
F6620.S027
CC6_4
zr6620_27V1V3_R1.fastq.gz
zr6620_27V1V3_R2.fastq.gz
F6620.S028
CC7_4
zr6620_28V1V3_R1.fastq.gz
zr6620_28V1V3_R2.fastq.gz
F6620.S029
CC8_4
zr6620_29V1V3_R1.fastq.gz
zr6620_29V1V3_R2.fastq.gz
F6620.S002
SupraC2
zr6620_2V1V3_R1.fastq.gz
zr6620_2V1V3_R2.fastq.gz
F6620.S030
CC9_4
zr6620_30V1V3_R1.fastq.gz
zr6620_30V1V3_R2.fastq.gz
F6620.S031
CC10_4
zr6620_31V1V3_R1.fastq.gz
zr6620_31V1V3_R2.fastq.gz
F6620.S032
FB1_11
zr6620_32V1V3_R1.fastq.gz
zr6620_32V1V3_R2.fastq.gz
F6620.S033
FB2_11
zr6620_33V1V3_R1.fastq.gz
zr6620_33V1V3_R2.fastq.gz
F6620.S034
FB3_11
zr6620_34V1V3_R1.fastq.gz
zr6620_34V1V3_R2.fastq.gz
F6620.S035
FA1_11
zr6620_35V1V3_R1.fastq.gz
zr6620_35V1V3_R2.fastq.gz
F6620.S036
FA2_11
zr6620_36V1V3_R1.fastq.gz
zr6620_36V1V3_R2.fastq.gz
F6620.S037
FA3_11
zr6620_37V1V3_R1.fastq.gz
zr6620_37V1V3_R2.fastq.gz
F6620.S038
B1_11
zr6620_38V1V3_R1.fastq.gz
zr6620_38V1V3_R2.fastq.gz
F6620.S039
B2_11
zr6620_39V1V3_R1.fastq.gz
zr6620_39V1V3_R2.fastq.gz
F6620.S003
SupraC3
zr6620_3V1V3_R1.fastq.gz
zr6620_3V1V3_R2.fastq.gz
F6620.S040
B3_11
zr6620_40V1V3_R1.fastq.gz
zr6620_40V1V3_R2.fastq.gz
F6620.S041
L1_11
zr6620_41V1V3_R1.fastq.gz
zr6620_41V1V3_R2.fastq.gz
F6620.S042
L2_11
zr6620_42V1V3_R1.fastq.gz
zr6620_42V1V3_R2.fastq.gz
F6620.S043
L3_11
zr6620_43V1V3_R1.fastq.gz
zr6620_43V1V3_R2.fastq.gz
F6620.S044
C1_11
zr6620_44V1V3_R1.fastq.gz
zr6620_44V1V3_R2.fastq.gz
F6620.S045
C2_11
zr6620_45V1V3_R1.fastq.gz
zr6620_45V1V3_R2.fastq.gz
F6620.S046
C3_11
zr6620_46V1V3_R1.fastq.gz
zr6620_46V1V3_R2.fastq.gz
F6620.S047
FB1_18
zr6620_47V1V3_R1.fastq.gz
zr6620_47V1V3_R2.fastq.gz
F6620.S048
FB2_18
zr6620_48V1V3_R1.fastq.gz
zr6620_48V1V3_R2.fastq.gz
F6620.S049
FB3_18
zr6620_49V1V3_R1.fastq.gz
zr6620_49V1V3_R2.fastq.gz
F6620.S004
SupraT1
zr6620_4V1V3_R1.fastq.gz
zr6620_4V1V3_R2.fastq.gz
F6620.S050
FA1_18
zr6620_50V1V3_R1.fastq.gz
zr6620_50V1V3_R2.fastq.gz
F6620.S051
FA2_18
zr6620_51V1V3_R1.fastq.gz
zr6620_51V1V3_R2.fastq.gz
F6620.S052
FA3_18
zr6620_52V1V3_R1.fastq.gz
zr6620_52V1V3_R2.fastq.gz
F6620.S053
B1_18
zr6620_53V1V3_R1.fastq.gz
zr6620_53V1V3_R2.fastq.gz
F6620.S054
B2_18
zr6620_54V1V3_R1.fastq.gz
zr6620_54V1V3_R2.fastq.gz
F6620.S055
B3_18
zr6620_55V1V3_R1.fastq.gz
zr6620_55V1V3_R2.fastq.gz
F6620.S056
L1_18
zr6620_56V1V3_R1.fastq.gz
zr6620_56V1V3_R2.fastq.gz
F6620.S057
L2_18
zr6620_57V1V3_R1.fastq.gz
zr6620_57V1V3_R2.fastq.gz
F6620.S058
L3_18
zr6620_58V1V3_R1.fastq.gz
zr6620_58V1V3_R2.fastq.gz
F6620.S059
C1_18
zr6620_59V1V3_R1.fastq.gz
zr6620_59V1V3_R2.fastq.gz
F6620.S005
SupraT2
zr6620_5V1V3_R1.fastq.gz
zr6620_5V1V3_R2.fastq.gz
F6620.S060
C2_18
zr6620_60V1V3_R1.fastq.gz
zr6620_60V1V3_R2.fastq.gz
F6620.S061
C3_18
zr6620_61V1V3_R1.fastq.gz
zr6620_61V1V3_R2.fastq.gz
F6620.S062
FB1_25
zr6620_62V1V3_R1.fastq.gz
zr6620_62V1V3_R2.fastq.gz
F6620.S063
FB2_25
zr6620_63V1V3_R1.fastq.gz
zr6620_63V1V3_R2.fastq.gz
F6620.S064
FB3_25
zr6620_64V1V3_R1.fastq.gz
zr6620_64V1V3_R2.fastq.gz
F6620.S065
FA1_25
zr6620_65V1V3_R1.fastq.gz
zr6620_65V1V3_R2.fastq.gz
F6620.S066
FA2_25
zr6620_66V1V3_R1.fastq.gz
zr6620_66V1V3_R2.fastq.gz
F6620.S067
FA3_25
zr6620_67V1V3_R1.fastq.gz
zr6620_67V1V3_R2.fastq.gz
F6620.S068
B1_25
zr6620_68V1V3_R1.fastq.gz
zr6620_68V1V3_R2.fastq.gz
F6620.S069
B2_25
zr6620_69V1V3_R1.fastq.gz
zr6620_69V1V3_R2.fastq.gz
F6620.S006
SupraT3
zr6620_6V1V3_R1.fastq.gz
zr6620_6V1V3_R2.fastq.gz
F6620.S070
B3_25
zr6620_70V1V3_R1.fastq.gz
zr6620_70V1V3_R2.fastq.gz
F6620.S071
L1_32
zr6620_71V1V3_R1.fastq.gz
zr6620_71V1V3_R2.fastq.gz
F6620.S072
L2_25
zr6620_72V1V3_R1.fastq.gz
zr6620_72V1V3_R2.fastq.gz
F6620.S073
L3_25
zr6620_73V1V3_R1.fastq.gz
zr6620_73V1V3_R2.fastq.gz
F6620.S074
C1_25
zr6620_74V1V3_R1.fastq.gz
zr6620_74V1V3_R2.fastq.gz
F6620.S075
C2_25
zr6620_75V1V3_R1.fastq.gz
zr6620_75V1V3_R2.fastq.gz
F6620.S076
C3_25
zr6620_76V1V3_R1.fastq.gz
zr6620_76V1V3_R2.fastq.gz
F6620.S077
FB1_32
zr6620_77V1V3_R1.fastq.gz
zr6620_77V1V3_R2.fastq.gz
F6620.S078
FB2_32
zr6620_78V1V3_R1.fastq.gz
zr6620_78V1V3_R2.fastq.gz
F6620.S079
FB3_32
zr6620_79V1V3_R1.fastq.gz
zr6620_79V1V3_R2.fastq.gz
F6620.S007
FB1_4
zr6620_7V1V3_R1.fastq.gz
zr6620_7V1V3_R2.fastq.gz
F6620.S080
FA1_32
zr6620_80V1V3_R1.fastq.gz
zr6620_80V1V3_R2.fastq.gz
F6620.S081
FA2_32
zr6620_81V1V3_R1.fastq.gz
zr6620_81V1V3_R2.fastq.gz
F6620.S082
FA3_32
zr6620_82V1V3_R1.fastq.gz
zr6620_82V1V3_R2.fastq.gz
F6620.S083
B1_32
zr6620_83V1V3_R1.fastq.gz
zr6620_83V1V3_R2.fastq.gz
F6620.S084
B2_32
zr6620_84V1V3_R1.fastq.gz
zr6620_84V1V3_R2.fastq.gz
F6620.S085
B3_32
zr6620_85V1V3_R1.fastq.gz
zr6620_85V1V3_R2.fastq.gz
F6620.S086
L1_25
zr6620_86V1V3_R1.fastq.gz
zr6620_86V1V3_R2.fastq.gz
F6620.S087
L2_32
zr6620_87V1V3_R1.fastq.gz
zr6620_87V1V3_R2.fastq.gz
F6620.S088
L3_32
zr6620_88V1V3_R1.fastq.gz
zr6620_88V1V3_R2.fastq.gz
F6620.S089
C1_32
zr6620_89V1V3_R1.fastq.gz
zr6620_89V1V3_R2.fastq.gz
F6620.S008
FB2_4
zr6620_8V1V3_R1.fastq.gz
zr6620_8V1V3_R2.fastq.gz
F6620.S090
C2_32
zr6620_90V1V3_R1.fastq.gz
zr6620_90V1V3_R2.fastq.gz
F6620.S091
C3_32
zr6620_91V1V3_R1.fastq.gz
zr6620_91V1V3_R2.fastq.gz
F6620.S092
FB1_39
zr6620_92V1V3_R1.fastq.gz
zr6620_92V1V3_R2.fastq.gz
F6620.S093
FB2_39
zr6620_93V1V3_R1.fastq.gz
zr6620_93V1V3_R2.fastq.gz
F6620.S094
FB3_39
zr6620_94V1V3_R1.fastq.gz
zr6620_94V1V3_R2.fastq.gz
F6620.S095
FA1_39
zr6620_95V1V3_R1.fastq.gz
zr6620_95V1V3_R2.fastq.gz
F6620.S096
FA2_39
zr6620_96V1V3_R1.fastq.gz
zr6620_96V1V3_R2.fastq.gz
F6620.S097
FA3_39
zr6620_97V1V3_R1.fastq.gz
zr6620_97V1V3_R2.fastq.gz
F6620.S098
B1_39
zr6620_98V1V3_R1.fastq.gz
zr6620_98V1V3_R2.fastq.gz
F6620.S099
B2_39
zr6620_99V1V3_R1.fastq.gz
zr6620_99V1V3_R2.fastq.gz
F6620.S009
FB3_4
zr6620_9V1V3_R1.fastq.gz
zr6620_9V1V3_R2.fastq.gz
Please download and save the file to your computer storage device. The download link will expire after 60 days upon your receiving of this report.
DADA2 is a software package that models and corrects Illumina-sequenced amplicon errors.
DADA2 infers sample sequences exactly, without coarse-graining into OTUs,
and resolves differences of as little as one nucleotide. DADA2 identified more real variants
and output fewer spurious sequences than other methods.
DADA2’s advantage is that it uses more of the data. The DADA2 error model incorporates quality information,
which is ignored by all other methods after filtering. The DADA2 error model incorporates quantitative abundances,
whereas most other methods use abundance ranks if they use abundance at all.
The DADA2 error model identifies the differences between sequences, eg. A->C,
whereas other methods merely count the mismatches. DADA2 can parameterize its error model from the data itself,
rather than relying on previous datasets that may or may not reflect the PCR and sequencing protocols used in your study.
DADA2 pipeline includes several tools for read quality control, including quality filtering, trimming, denoising, pair merging and chimera filtering. Below are the major processing steps of DADA2:
Step 1. Read trimming based on sequence quality
The quality of NGS Illumina sequences often decreases toward the end of the reads.
DADA2 allows to trim off the poor quality read ends in order to improve the error
model building and pair mergicing performance.
Step 2. Learn the Error Rates
The DADA2 algorithm makes use of a parametric error model (err) and every
amplicon dataset has a different set of error rates. The learnErrors method
learns this error model from the data, by alternating estimation of the error
rates and inference of sample composition until they converge on a jointly
consistent solution. As in many machine-learning problems, the algorithm must
begin with an initial guess, for which the maximum possible error rates in
this data are used (the error rates if only the most abundant sequence is
correct and all the rest are errors).
Step 3. Infer amplicon sequence variants (ASVs) based on the error model built in previous step. This step is also called sequence "denoising".
The outcome of this step is a list of ASVs that are the equivalent of oligonucleotides.
Step 4. Merge paired reads. If the sequencing products are read pairs, DADA2 will merge the R1 and R2 ASVs into single sequences.
Merging is performed by aligning the denoised forward reads with the reverse-complement of the corresponding
denoised reverse reads, and then constructing the merged “contig” sequences.
By default, merged sequences are only output if the forward and reverse reads overlap by
at least 12 bases, and are identical to each other in the overlap region (but these conditions can be changed via function arguments).
Step 5. Remove chimera.
The core dada method corrects substitution and indel errors, but chimeras remain. Fortunately, the accuracy of sequence variants
after denoising makes identifying chimeric ASVs simpler than when dealing with fuzzy OTUs.
Chimeric sequences are identified if they can be exactly reconstructed by
combining a left-segment and a right-segment from two more abundant “parent” sequences. The frequency of chimeric sequences varies substantially
from dataset to dataset, and depends on on factors including experimental procedures and sample complexity.
Results
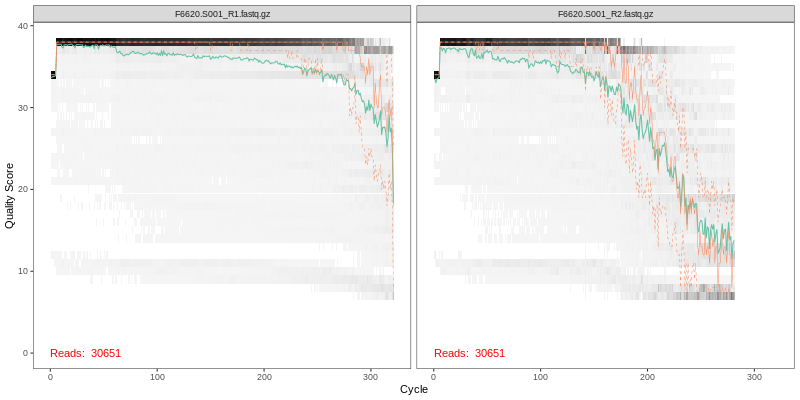
1. Read Quality Plots NGS sequence analaysis starts with visualizing the quality of the sequencing. Below are the quality plots of the first
sample for the R1 and R2 reads separately. In gray-scale is a heat map of the frequency of each quality score at each base position. The mean
quality score at each position is shown by the green line, and the quartiles of the quality score distribution by the orange lines.
The forward reads are usually of better quality. It is a common practice to trim the last few nucleotides to avoid less well-controlled errors
that can arise there. The trimming affects the downstream steps including error model building, merging and chimera calling. FOMC uses an empirical
approach to test many combinations of different trim length in order to achieve best final amplicon sequence variants (ASVs), see the next
section “Optimal trim length for ASVs”.
2. Optimal trim length for ASVs The final number of merged and chimera-filtered ASVs depends on the quality filtering (hence trimming) in the very beginning of the DADA2 pipeline.
In order to achieve highest number of ASVs, an empirical approach was used -
Create a random subset of each sample consisting of 5,000 R1 and 5,000 R2 (to reduce computation time)
Trim 10 bases at a time from the ends of both R1 and R2 up to 50 bases
For each combination of trimmed length (e.g., 300x300, 300x290, 290x290 etc), the trimmed reads are
subject to the entire DADA2 pipeline for chimera-filtered merged ASVs
The combination with highest percentage of the input reads becoming final ASVs is selected for the complete set of data
Below is the result of such operation, showing ASV percentages of total reads for all trimming combinations (1st Column = R1 lengths in bases; 1st Row = R2 lengths in bases):
R1/R2
281
271
261
251
241
231
221
211
201
321
26.04%
47.96%
56.00%
60.47%
60.32%
34.52%
28.11%
19.18%
8.92%
311
26.16%
49.63%
58.20%
62.28%
35.37%
29.15%
21.77%
11.17%
4.37%
301
26.44%
49.98%
57.84%
35.61%
28.53%
20.15%
11.00%
4.19%
3.64%
291
26.24%
49.84%
33.74%
28.22%
19.88%
10.86%
4.10%
3.44%
0.02%
281
29.91%
29.90%
27.85%
19.88%
10.69%
4.16%
3.53%
0.02%
0.02%
271
7.19%
23.89%
18.82%
10.03%
4.08%
3.50%
0.02%
0.02%
0.00%
261
3.90%
15.69%
9.32%
3.80%
3.52%
0.02%
0.02%
0.00%
0.00%
251
2.73%
8.32%
3.54%
3.36%
0.02%
0.02%
0.00%
0.00%
0.00%
241
0.53%
3.69%
3.50%
0.02%
0.02%
0.00%
0.00%
0.00%
0.00%
Based on the above result, the trim length combination of R1 = 311 bases and R2 = 251 bases (highlighted red above), was chosen for generating final ASVs for all sequences.
This combination generated highest number of merged non-chimeric ASVs and was used for downstream analyses, if requested.
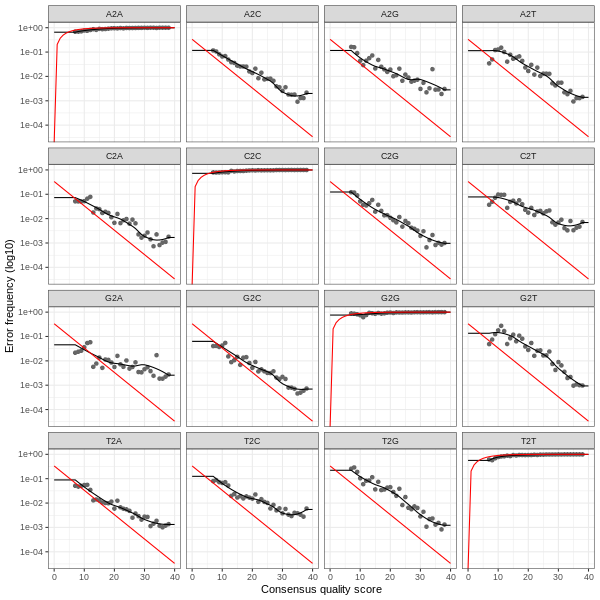
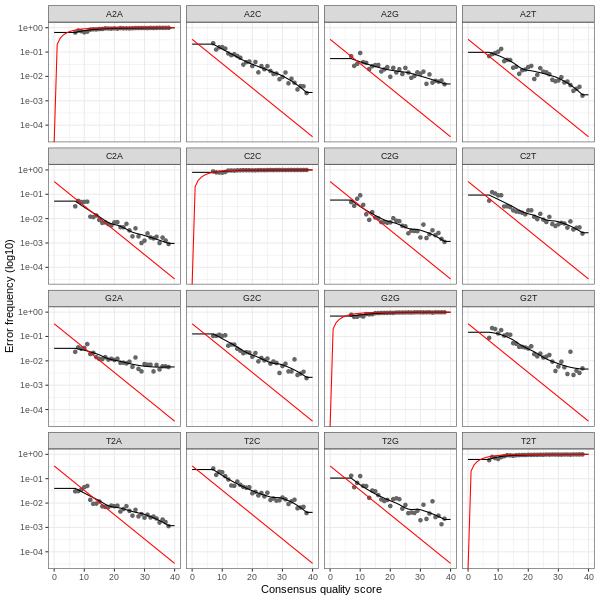
3. Error plots from learning the error rates
After DADA2 building the error model for the set of data, it is always worthwhile, as a sanity check if nothing else, to visualize the estimated error rates.
The error rates for each possible transition (A→C, A→G, …) are shown below. Points are the observed error rates for each consensus quality score.
The black line shows the estimated error rates after convergence of the machine-learning algorithm.
The red line shows the error rates expected under the nominal definition of the Q-score.
The ideal result would be the estimated error rates (black line) are a good fit to the observed rates (points), and the error rates drop
with increased quality as expected.
Forward Read R1 Error Plot
Reverse Read R2 Error Plot
The PDF version of these plots are available here:
4. DADA2 Result Summary The table below shows the summary of the DADA2 analysis,
tracking paired read counts of each samples for all the steps during DADA2 denoising process -
including end-trimming (filtered), denoising (denoisedF, denoisedF), pair merging (merged) and chimera removal (nonchim).
Sample ID
F6620.S001
F6620.S002
F6620.S003
F6620.S004
F6620.S005
F6620.S006
F6620.S007
F6620.S008
F6620.S009
F6620.S010
F6620.S011
F6620.S012
F6620.S013
F6620.S014
F6620.S015
F6620.S016
F6620.S017
F6620.S018
F6620.S019
F6620.S020
F6620.S021
F6620.S022
F6620.S023
F6620.S024
F6620.S025
F6620.S026
F6620.S027
F6620.S028
F6620.S029
F6620.S030
F6620.S031
F6620.S032
F6620.S033
F6620.S034
F6620.S035
F6620.S036
F6620.S037
F6620.S038
F6620.S039
F6620.S040
F6620.S041
F6620.S042
F6620.S043
F6620.S044
F6620.S045
F6620.S046
F6620.S047
F6620.S048
F6620.S049
F6620.S050
F6620.S051
F6620.S052
F6620.S053
F6620.S054
F6620.S055
F6620.S056
F6620.S057
F6620.S058
F6620.S059
F6620.S060
F6620.S061
F6620.S062
F6620.S063
F6620.S064
F6620.S065
F6620.S066
F6620.S067
F6620.S068
F6620.S069
F6620.S070
F6620.S071
F6620.S072
F6620.S073
F6620.S074
F6620.S075
F6620.S076
F6620.S077
F6620.S078
F6620.S079
F6620.S080
F6620.S081
F6620.S082
F6620.S083
F6620.S084
F6620.S085
F6620.S086
F6620.S087
F6620.S088
F6620.S089
F6620.S090
F6620.S091
F6620.S092
F6620.S093
F6620.S094
F6620.S095
F6620.S096
F6620.S097
F6620.S098
F6620.S099
F6620.S100
F6620.S101
F6620.S102
F6620.S103
F6620.S104
F6620.S105
F6620.S106
F6620.S107
F6620.S108
F6620.S109
F6620.S110
F6620.S111
F6620.S112
F6620.S113
F6620.S114
F6620.S115
F6620.S116
F6620.S117
F6620.S118
F6620.S119
F6620.S120
F6620.S121
F6620.S122
F6620.S123
F6620.S124
F6620.S125
F6620.S126
F6620.S127
F6620.S128
F6620.S129
F6620.S130
F6620.S131
F6620.S132
F6620.S133
F6620.S134
F6620.S135
F6620.S136
F6620.S137
F6620.S138
F6620.S139
Row Sum
Percentage
input
30,651
35,995
27,863
30,085
35,907
25,324
19,473
29,904
31,956
31,109
30,750
28,393
33,816
36,335
33,000
35,645
31,858
31,931
35,239
32,490
33,843
37,936
31,752
35,829
43,437
35,200
35,585
36,355
34,803
28,498
25,177
31,251
38,276
30,610
30,955
23,338
27,360
31,329
35,992
46,258
33,195
36,033
29,205
28,231
39,444
41,980
31,442
31,710
27,748
31,592
27,096
28,485
42,189
38,303
29,690
30,071
39,132
33,166
42,444
23,135
29,357
32,558
33,866
35,246
31,788
23,440
27,888
33,026
26,136
28,982
29,474
28,431
22,610
30,522
32,564
23,166
28,288
28,825
31,834
25,117
34,245
29,669
19,086
28,235
36,390
26,800
29,989
28,027
37,281
30,414
25,310
26,974
28,201
30,714
23,890
35,835
33,775
29,357
33,034
30,698
27,542
26,729
28,828
27,480
34,728
21,781
32,048
34,599
34,512
26,012
29,609
28,027
36,847
33,047
27,707
32,213
27,275
29,983
23,064
28,967
31,435
36,930
31,813
27,328
22,984
39,409
20,519
30,119
41,051
31,677
27,843
27,643
32,022
26,810
32,621
22,097
28,041
40,678
29,799
4,318,658
100.00%
filtered
30,645
35,968
27,841
30,061
35,891
25,300
19,464
29,890
31,926
31,093
30,725
28,370
33,798
36,307
32,986
35,630
31,829
31,913
35,216
32,462
33,812
37,901
31,732
35,816
43,409
35,176
35,560
36,339
34,775
28,484
25,166
31,239
38,251
30,584
30,943
23,321
27,350
31,303
35,967
46,243
33,175
36,013
29,185
28,214
39,418
41,951
31,418
31,686
27,723
31,581
27,088
28,468
42,167
38,282
29,670
30,054
39,098
33,148
42,413
23,124
29,338
32,535
33,842
35,226
31,770
23,424
27,866
33,009
26,123
28,969
29,463
28,409
22,591
30,506
32,545
23,144
28,270
28,802
31,820
25,104
34,219
29,652
19,074
28,224
36,368
26,786
29,980
28,016
37,262
30,398
25,298
26,945
28,182
30,689
23,863
35,827
33,749
29,337
33,010
30,666
27,499
26,712
28,818
27,461
34,702
21,762
32,027
34,585
34,500
25,982
29,594
28,011
36,825
33,031
27,691
32,193
27,255
29,958
23,050
28,936
31,402
36,903
31,798
27,314
22,980
39,374
20,510
30,109
41,019
31,660
27,820
27,630
31,998
26,799
32,601
22,087
28,022
40,645
29,774
4,315,900
99.94%
denoisedF
28,764
33,780
25,927
28,047
33,769
23,460
18,267
28,264
29,969
28,966
28,662
26,233
31,442
33,903
31,035
33,760
29,938
29,612
33,606
30,952
31,840
36,253
30,274
34,317
41,396
33,451
33,885
34,613
33,042
27,201
23,922
29,949
36,361
29,011
29,096
21,914
25,560
29,898
33,987
43,867
31,292
34,156
27,534
26,683
37,331
39,870
30,121
30,144
26,554
30,383
25,825
26,890
40,736
36,616
28,166
28,720
37,468
31,629
40,823
21,986
27,872
31,357
32,466
33,968
30,454
22,426
26,496
31,871
25,058
27,732
27,858
26,919
21,526
29,166
31,047
21,892
26,796
27,574
30,547
24,074
32,590
28,361
18,260
26,879
34,928
25,592
28,856
26,810
35,567
29,120
24,177
25,636
27,001
29,319
22,602
34,315
32,419
28,047
31,461
29,214
26,405
25,315
27,373
26,077
33,054
20,518
30,776
33,133
33,254
24,407
28,067
26,827
35,469
31,600
26,482
30,838
26,035
28,310
22,005
27,523
29,791
35,414
30,499
26,398
21,977
37,824
19,606
28,712
39,249
30,594
26,541
26,523
30,725
25,629
31,139
20,920
26,622
38,727
28,225
4,107,956
95.12%
denoisedR
27,852
32,398
25,100
27,481
32,772
22,904
17,932
27,483
29,654
28,203
28,336
25,940
31,384
33,378
30,199
33,085
29,493
29,618
33,046
29,751
30,774
34,016
29,826
33,826
40,472
32,864
33,424
34,202
32,589
26,696
23,676
28,911
35,995
27,941
28,325
21,414
24,953
28,917
33,494
43,175
31,005
33,365
27,396
26,028
36,686
39,062
29,302
29,749
25,118
29,504
24,999
26,280
39,482
35,676
27,721
28,617
36,880
30,904
39,922
21,335
26,791
29,716
31,730
33,299
29,868
21,645
26,285
30,661
24,028
27,129
27,383
26,791
20,021
28,531
29,229
21,237
26,296
26,968
29,646
23,042
31,914
27,532
17,309
26,105
33,803
24,828
28,206
25,951
35,204
28,646
23,569
25,003
25,715
28,748
22,103
33,675
31,569
26,664
30,772
28,794
26,151
24,837
26,851
25,531
32,373
19,242
29,890
32,198
32,333
24,301
27,867
25,883
33,235
30,236
25,731
30,221
25,083
27,897
21,226
26,543
29,310
34,591
29,452
25,600
21,179
36,643
19,208
27,716
38,458
29,139
26,002
25,714
29,399
24,796
30,194
20,217
25,711
38,235
27,137
4,003,266
92.70%
merged
23,703
27,741
21,039
22,660
27,362
17,981
15,398
23,664
25,541
23,127
23,350
21,133
25,732
27,440
24,735
27,802
25,094
24,825
27,599
24,688
26,137
29,298
25,820
28,202
32,240
27,895
27,436
28,809
26,854
23,268
20,802
25,268
31,608
25,279
24,861
18,163
21,400
26,171
29,785
37,595
26,116
28,442
23,307
22,134
31,604
32,836
25,704
26,255
22,263
25,489
22,159
23,467
34,915
31,768
22,847
25,438
32,961
27,991
35,531
19,178
24,144
27,492
28,319
29,829
25,982
19,566
22,776
28,986
22,734
24,357
24,157
23,132
17,734
25,478
25,750
17,893
23,176
23,980
26,558
20,259
27,775
23,462
15,204
23,014
29,861
22,064
25,201
23,281
30,570
25,159
21,211
22,401
23,232
25,464
19,456
29,291
27,649
23,746
27,982
26,070
23,615
21,986
23,440
22,386
27,627
17,094
27,160
29,086
28,910
20,778
24,064
23,133
29,957
26,841
23,937
26,626
22,450
24,964
16,395
22,551
25,793
31,409
26,806
23,538
18,848
31,945
17,037
25,354
36,417
27,744
23,578
23,209
26,557
21,679
27,525
18,245
22,385
33,024
23,075
3,499,473
81.03%
nonchim
14,217
16,130
12,874
13,678
16,451
10,507
12,287
17,708
19,421
15,776
15,639
14,746
17,606
18,068
16,505
17,657
16,557
15,639
19,833
18,654
18,710
20,380
17,134
19,406
22,379
19,616
19,817
19,397
15,992
13,983
14,628
21,032
24,346
20,177
20,113
14,698
16,958
18,506
20,711
26,564
18,864
19,519
16,428
15,700
23,004
22,768
19,536
20,340
18,151
19,815
17,660
18,643
25,188
23,292
18,329
18,278
23,491
19,838
25,500
15,113
18,817
22,614
22,387
24,677
21,276
15,048
16,964
18,419
14,280
16,800
17,855
18,045
13,467
20,973
19,936
11,998
19,536
19,253
23,068
17,348
21,219
17,597
12,787
17,531
21,848
16,857
20,126
17,745
23,428
18,030
15,755
17,644
18,293
22,114
17,183
22,560
22,947
17,725
19,310
18,828
18,211
17,471
17,781
15,814
19,571
12,756
21,375
22,597
25,441
16,919
19,369
19,641
22,253
19,260
17,105
21,989
18,606
20,429
13,030
16,637
19,391
24,282
20,407
20,824
15,370
24,326
13,680
16,444
23,415
17,786
19,333
19,734
20,790
15,625
18,894
13,918
16,256
23,310
16,896
2,593,181
60.05%
This table can be downloaded as an Excel table below:
5. DADA2 Amplicon Sequence Variants (ASVs). A total of 6177 unique merged and chimera-free ASV sequences were identified, and their corresponding
read counts for each sample are available in the "ASV Read Count Table" with rows for the ASV sequences and columns for sample. This read count table can be used for
microbial profile comparison among different samples and the sequences provided in the table can be used to taxonomy assignment.
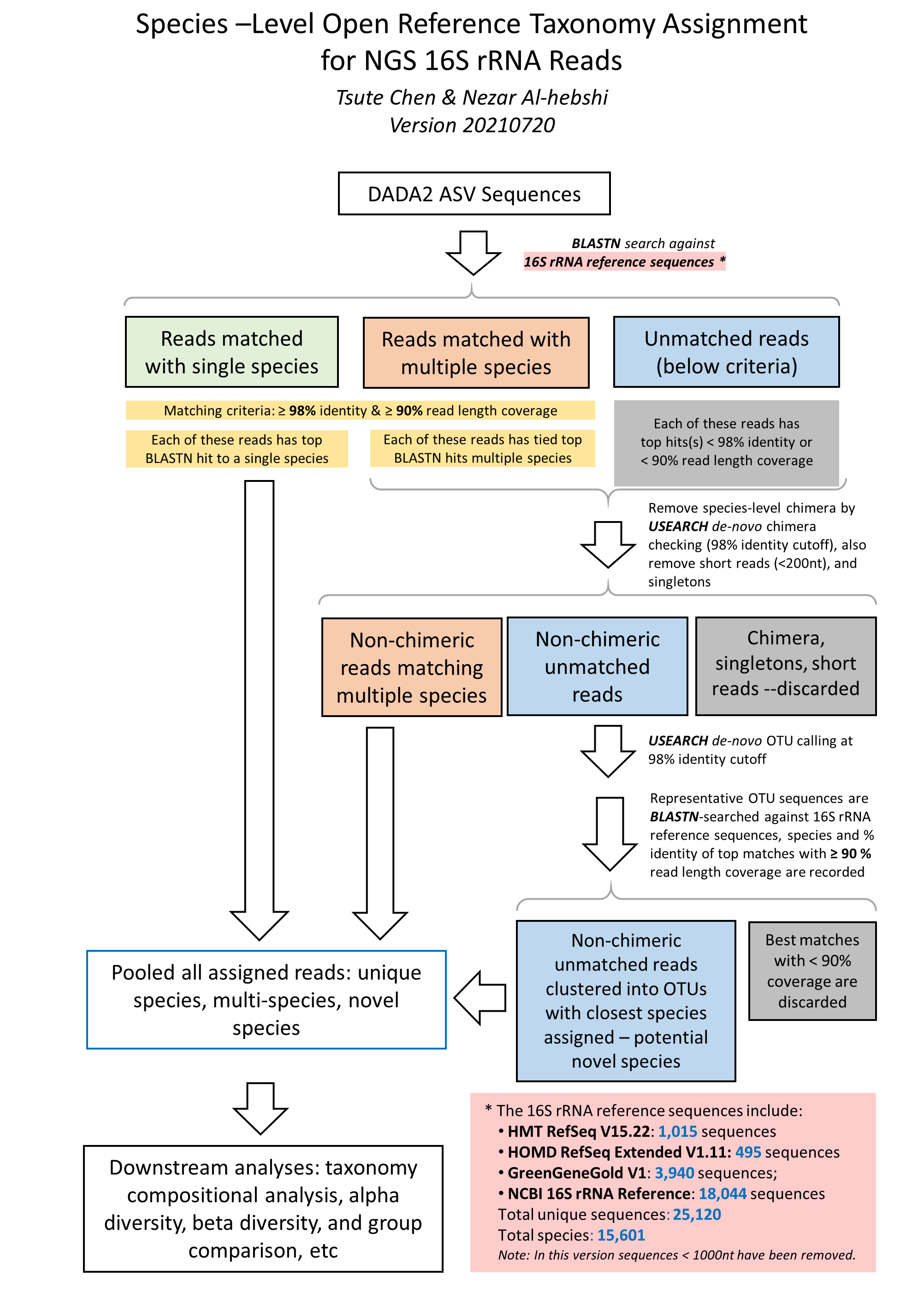
The species-level, open-reference 16S rRNA NGS reads taxonomy assignment pipeline
Version 20210310
1. Raw sequences reads in FASTA format were BLASTN-searched against a combined set of 16S rRNA reference sequences.
It consists of MOMD (version 0.1), the HOMD (version 15.2 http://www.homd.org/index.php?name=seqDownload&file&type=R ),
HOMD 16S rRNA RefSeq Extended Version 1.1 (EXT), GreenGene Gold (GG)
(http://greengenes.lbl.gov/Download/Sequence_Data/Fasta_data_files/gold_strains_gg16S_aligned.fasta.gz) ,
and the NCBI 16S rRNA reference sequence set (https://ftp.ncbi.nlm.nih.gov/blast/db/16S_ribosomal_RNA.tar.gz).
These sequences were screened and combined to remove short sequences (<1000nt), chimera, duplicated and sub-sequences,
as well as sequences with poor taxonomy annotation (e.g., without species information).
This process resulted in 1,015 from HOMD V15.22, 495 from EXT, 3,940 from GG and 18,044 from NCBI, a total of 25,120 sequences.
Altogether these sequence represent a total of 15,601 oral and non-oral microbial species.
The NCBI BLASTN version 2.7.1+ (Zhang et al, 2000) was used with the default parameters.
Reads with ≥ 98% sequence identity to the matched reference and ≥ 90% alignment length
(i.e., ≥ 90% of the read length that was aligned to the reference and was used to calculate
the sequence percent identity) were classified based on the taxonomy of the reference sequence
with highest sequence identity. If a read matched with reference sequences representing
more than one species with equal percent identity and alignment length, it was subject
to chimera checking with USEARCH program version v8.1.1861 (Edgar 2010). Non-chimeric reads with multi-species
best hits were considered valid and were assigned with a unique species
notation (e.g., spp) denoting unresolvable multiple species.
2. Unassigned reads (i.e., reads with < 98% identity or < 90% alignment length) were pooled together and reads < 200 bases were
removed. The remaining reads were subject to the de novo
operational taxonomy unit (OTU) calling and chimera checking using the USEARCH program version v8.1.1861 (Edgar 2010).
The de novo OTU calling and chimera checking was done using 98% as the sequence identity cutoff, i.e., the species-level OTU.
The output of this step produced species-level de novo clustered OTUs with 98% identity.
Representative reads from each of the OTUs/species were then BLASTN-searched
against the same reference sequence set again to determine the closest species for
these potential novel species. These potential novel species were pooled together with the reads that were signed to specie-level in
the previous step, for down-stream analyses.
Reference:
Edgar RC. Search and clustering orders of magnitude faster than BLAST.
Bioinformatics. 2010 Oct 1;26(19):2460-1. doi: 10.1093/bioinformatics/btq461. Epub 2010 Aug 12. PubMed PMID: 20709691.
3. Designations used in the taxonomy:
1) Taxonomy levels are indicated by these prefixes:
k__: domain/kingdom
p__: phylum
c__: class
o__: order
f__: family
g__: genus
s__: species
Example:
k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Blautia;s__faecis
2) Unique level identified – known species:
k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Roseburia;s__hominis
The above example shows some reads match to a single species (all levels are unique)
3) Non-unique level identified – known species:
k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Roseburia;s__multispecies_spp123_3
The above example “s__multispecies_spp123_3” indicates certain reads equally match to 3 species of the
genus Roseburia; the “spp123” is a temporally assigned species ID.
k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__multigenus;s__multispecies_spp234_5
The above example indicates certain reads match equally to 5 different species, which belong to multiple genera.;
the “spp234” is a temporally assigned species ID.
4) Unique level identified – unknown species, potential novel species:
k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Roseburia;s__ hominis_nov_97%
The above example indicates that some reads have no match to any of the reference sequences with
sequence identity ≥ 98% and percent coverage (alignment length) ≥ 98% as well. However this groups
of reads (actually the representative read from a de novo OTU) has 96% percent identity to
Roseburia hominis, thus this is a potential novel species, closest to Roseburia hominis.
(But they are not the same species).
5) Multiple level identified – unknown species, potential novel species:
k__Bacteria;p__Firmicutes;c__Clostridia;o__Clostridiales;f__Lachnospiraceae;g__Roseburia;s__ multispecies_sppn123_3_nov_96%
The above example indicates that some reads have no match to any of the reference sequences
with sequence identity ≥ 98% and percent coverage (alignment length) ≥ 98% as well.
However this groups of reads (actually the representative read from a de novo OTU)
has 96% percent identity equally to 3 species in Roseburia. Thus this is no single
closest species, instead this group of reads match equally to multiple species at 96%.
Since they have passed chimera check so they represent a novel species. “sppn123” is a
temporary ID for this potential novel species.
4. The taxonomy assignment algorithm is illustrated in this flow char below:
Read Taxonomy Assignment - Result Summary *
Code
Category
MPC=0% (>=1 read)
MPC=0.01%(>=259 reads)
A
Total reads
2,593,181
2,593,181
B
Total assigned reads
2,591,858
2,591,858
C
Assigned reads in species with read count < MPC
0
9,395
D
Assigned reads in samples with read count < 500
0
0
E
Total samples
139
139
F
Samples with reads >= 500
139
139
G
Samples with reads < 500
0
0
H
Total assigned reads used for analysis (B-C-D)
2,591,858
2,582,463
I
Reads assigned to single species
2,493,504
2,487,764
J
Reads assigned to multiple species
90,915
90,564
K
Reads assigned to novel species
7,439
4,135
L
Total number of species
307
144
M
Number of single species
204
129
N
Number of multi-species
12
7
O
Number of novel species
91
8
P
Total unassigned reads
1,323
1,323
Q
Chimeric reads
23
23
R
Reads without BLASTN hits
51
51
S
Others: short, low quality, singletons, etc.
1,249
1,249
A=B+P=C+D+H+Q+R+S
E=F+G
B=C+D+H
H=I+J+K
L=M+N+O
P=Q+R+S
* MPC = Minimal percent (of all assigned reads) read count per species, species with read count < MPC were removed.
* Samples with reads < 500 were removed from downstream analyses.
* The assignment result from MPC=0.1% was used in the downstream analyses.
Read Taxonomy Assignment - ASV Species-Level Read Counts Table
This table shows the read counts for each sample (columns) and each species identified based on the ASV sequences.
The downstream analyses were based on this table.
The species listed in the table has full taxonomy and a dynamically assigned species ID specific to this report.
When some reads match with the reference sequences of more than one species equally (i.e., same percent identiy and alignmnet coverage),
they can't be assigned to a particular species. Instead, they are assigned to multiple species with the species notaton
"s__multispecies_spp2_2". In this notation, spp2 is the dynamic ID assigned to these reads that hit multiple sequences and the "_2"
at the end of the notation means there are two species in the spp2.
You can look up which species are included in the multi-species assignment, in this table below:
Another type of notation is "s__multispecies_sppn2_2", in which the "n" in the sppn2 means it's a potential novel species because all the reads in this species
have < 98% idenity to any of the reference sequences. They were grouped together based on de novo OTU clustering at 98% identity cutoff. And then
a representative sequence was chosed to BLASTN search against the reference database to find the closest match (but will still be < 98%). This representative
sequence also matched equally to more than one species, hence the "spp" was given in the label.
In ecology, alpha diversity (α-diversity) is the mean species diversity in sites or habitats at a local scale.
The term was introduced by R. H. Whittaker[1][2] together with the terms beta diversity (β-diversity)
and gamma diversity (γ-diversity). Whittaker's idea was that the total species diversity in a landscape
(gamma diversity) is determined by two different things, the mean species diversity in sites or habitats
at a more local scale (alpha diversity) and the differentiation among those habitats (beta diversity).
Diversity measures are affected by the sampling depth. Rarefaction is a technique to assess species richness from the results of sampling. Rarefaction allows
the calculation of species richness for a given number of individual samples, based on the construction
of so-called rarefaction curves. This curve is a plot of the number of species as a function of the
number of samples. Rarefaction curves generally grow rapidly at first, as the most common species are found,
but the curves plateau as only the rarest species remain to be sampled.
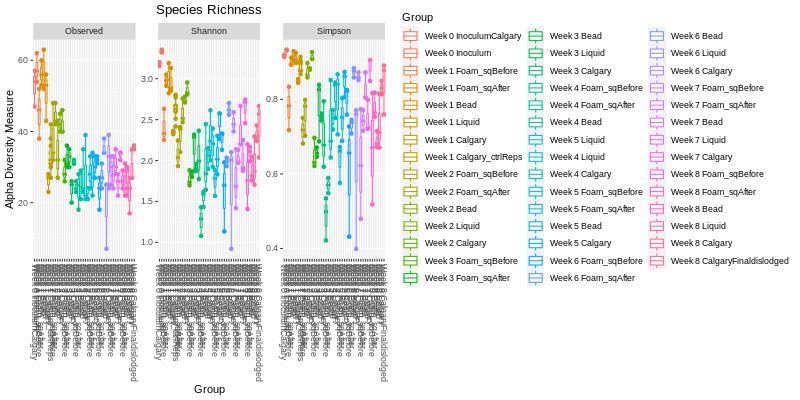
The two main factors taken into account when measuring diversity are richness and evenness.
Richness is a measure of the number of different kinds of organisms present in a particular area.
Evenness compares the similarity of the population size of each of the species present. There are
many different ways to measure the richness and evenness. These measurements are called "estimators" or "indices".
Below is a diversity of 3 commonly used indices showing the values for all the samples (dots) and in groups (boxes).
Alpha Diversity Box Plots for All Groups
Alpha Diversity Box Plots for Individual Comparisons
To test whether the alpha diversity among different comparison groups are different statisticall, we use the Kruskal Wallis H test
provided the "alpha-group-significance" fucntion in the QIIME 2 diversity package. Kruskal Wallis H test is the non parametric alternative
to the One Way ANOVA. Non parametric means that the test doesn’t assume your data comes from a particular distribution. The H test is used
when the assumptions for ANOVA aren’t met (like the assumption of normality). It is sometimes called the one-way ANOVA on ranks,
as the ranks of the data values are used in the test rather than the actual data points. The test determines whether the medians of two
or more groups are different.
Below are the Kruskal Wallis H test results for each comparison based on three different alpha diversity measures: 1) Observed species (features),
2) Shannon index, and 3) Simpson index.
Beta diversity compares the similarity (or dissimilarity) of microbial profiles between different
groups of samples. There are many different similarity/dissimilarity metrics.
In general, they can be quantitative (using sequence abundance, e.g., Bray-Curtis or weighted UniFrac)
or binary (considering only presence-absence of sequences, e.g., binary Jaccard or unweighted UniFrac).
They can be even based on phylogeny (e.g., UniFrac metrics) or not (non-UniFrac metrics, such as Bray-Curtis, etc.).
For microbiome studies, species profiles of samples can be compared with the Bray-Curtis dissimilarity,
which is based on the count data type. The pair-wise Bray-Curtis dissimilarity matrix of all samples can then be
subject to either multi-dimensional scaling (MDS, also known as PCoA) or non-metric MDS (NMDS).
MDS/PCoA is a
scaling or ordination method that starts with a matrix of similarities or dissimilarities
between a set of samples and aims to produce a low-dimensional graphical plot of the data
in such a way that distances between points in the plot are close to original dissimilarities.
NMDS is similar to MDS, however it does not use the dissimilarities data, instead it converts them into
the ranks and use these ranks in the calculation.
In our beta diversity analysis, Bray-Curtis dissimilarity matrix was first calculated and then plotted by the PCoA and
NMDS separately. Below are beta diveristy results for all groups together:
NMDS and PCoA Plots for All Groups
The above PCoA and NMDS plots are based on count data. The count data can also be transformed into centered log ratio (CLR)
for each species. The CLR data is no longer count data and cannot be used in Bray-Curtis dissimilarity calculation. Instead
CLR can be compared with Euclidean distances. When CLR data are compared by Euclidean distance, the distance is also called
Aitchison distance.
Below are the NMDS and PCoA plots of the Aitchison distances of the samples:
Interactive 3D PCoA Plots - Bray-Curtis Dissimilarity
Interactive 3D PCoA Plots - Euclidean Distance
Interactive 3D PCoA Plots - Correlation Coefficients
Group Significance of Beta-diversity Indices
To test whether the between-group dissimilarities are significantly greater than the within-group dissimilarities,
the "beta-group-significance" function provided in the QIIME 2 "diversity" package was used with PERMANOVA
(permutational multivariate analysis of variance) chosen s the group significan testing method.
Three beta diversity matrics were used: 1) Bray–Curtis dissimilarity 2) Correlation coefficient matrix , and 3) Aitchison distance
(Euclidean distance between clr-transformed compositions).
16S rRNA next generation sequencing (NGS) generates a fixed number of reads that reflect the proportion of different
species in a sample, i.e., the relative abundance of species, instead of the absolute abundance.
In Mathematics, measurements involving probabilities, proportions, percentages, and ppm can all
be thought of as compositional data. This makes the microbiome read count data “compositional”
(Gloor et al, 2017). In general, compositional data represent parts of a whole which only
carry relative information (http://www.compositionaldata.com/).
The problem of microbiome data being compositional arises when comparing two groups of samples for
identifying “differentially abundant” species. A species with the same absolute abundance between two
conditions, its relative abundances in the two conditions (e.g., percent abundance) can become different
if the relative abundance of other species change greatly. This problem can lead to incorrect conclusion
in terms of differential abundance for microbial species in the samples.
When studying differential abundance (DA), the current better approach is to transform the read count
data into log ratio data. The ratios are calculated between read counts of all species in a sample to
a “reference” count (e.g., mean read count of the sample). The log ratio data allow the detection of DA
species without being affected by percentage bias mentioned above
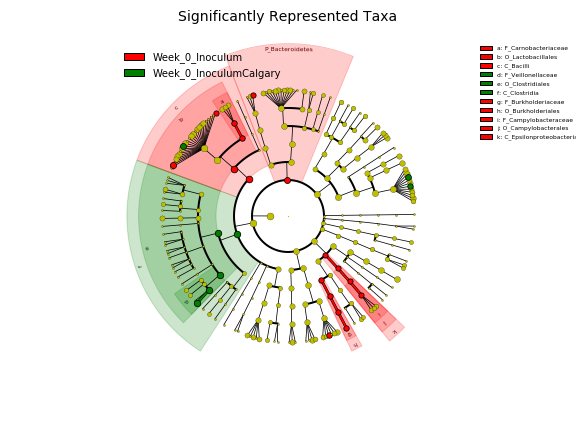
In this report, a compositional DA analysis tool “ANCOM” (analysis of composition of microbiomes)
was used. ANCOM transforms the count data into log-ratios and thus is more suitable for comparing
the composition of microbiomes in two or more populations. "ANCOM" generates a table of features with
W-statistics and whether the null hypothesis is rejected. The “W” is the W-statistic, or number of
features that a single feature is tested to be significantly different against. Hence the higher the "W"
the more statistical sifgnificane that a feature/species is differentially abundant.
References:
Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome Datasets Are Compositional: And This Is Not Optional. Front Microbiol.
2017 Nov 15;8:2224. doi: 10.3389/fmicb.2017.02224. PMID: 29187837; PMCID: PMC5695134.
Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. Analysis of composition of
microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis.
2015 May 29;26:27663. doi: 10.3402/mehd.v26.27663. PMID: 26028277; PMCID: PMC4450248.
Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction.
Nat Commun. 2020 Jul 14;11(1):3514. doi: 10.1038/s41467-020-17041-7.
PMID: 32665548; PMCID: PMC7360769.
Starting with version V1.2, we also include the results of ANCOM-BC (Analysis of Compositions of
Microbiomes with Bias Correction) (Lin and Peddada 2020). ANCOM-BC is an updated version of "ANCOM" that:
(a) provides statistically valid test with appropriate p-values,
(b) provides confidence intervals for differential abundance of each taxon,
(c) controls the False Discovery Rate (FDR),
(d) maintains adequate power, and
(e) is computationally simple to implement.
The bias correction (BC) addresses a challenging problem of the bias introduced by differences in
the sampling fractions across samples. This bias has been a major hurdle in performing DA analysis of microbiome data.
ANCOM-BC estimates the unknown sampling fractions and corrects the bias induced by their differences among samples.
The absolute abundance data are modeled using a linear regression framework.
References:
Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction.
Nat Commun. 2020 Jul 14;11(1):3514. doi: 10.1038/s41467-020-17041-7.
PMID: 32665548; PMCID: PMC7360769.
LEfSe (Linear Discriminant Analysis Effect Size) is an alternative method to find "organisms, genes, or
pathways that consistently explain the differences between two or more microbial communities" (Segata et al., 2011).
Specifically, LEfSe uses rank-based Kruskal-Wallis (KW) sum-rank test to detect features with significant
differential (relative) abundance with respect to the class of interest. Since it is rank-based, instead of proportional based,
the differential species identified among the comparison groups is less biased (than percent abundance based).
Reference:
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011 Jun 24;12(6):R60. doi: 10.1186/gb-2011-12-6-r60. PMID: 21702898; PMCID: PMC3218848.
To analyze the co-occurrence or co-exclusion between microbial species among different samples, network correlation
analysis tools are usually used for this purpose. However, microbiome count data are compositional. If count data are normalized to the total number of counts in the
sample, the data become not independent and traditional statistical metrics (e.g., correlation) for the detection
of specie-species relationships can lead to spurious results. In addition, sequencing-based studies typically
measure hundreds of OTUs (species) on few samples; thus, inference of OTU-OTU association networks is severely
under-powered. Here we use SPIEC-EASI (SParse InversECovariance Estimation
for Ecological Association Inference), a statistical method for the inference of microbial
ecological networks from amplicon sequencing datasets that addresses both of these issues (Kurtz et al., 2015).
SPIEC-EASI combines data transformations developed for compositional data analysis with a graphical model
inference framework that assumes the underlying ecological association network is sparse. SPIEC-EASI provides
two algorithms for network inferencing – 1) Meinshausen-Bühlmann's neighborhood selection (MB method) and inverse covariance selection
(GLASSO method, i.e., graphical least absolute shrinkage and selection operator). This is fundamentally distinct from SparCC, which essentially estimate pairwise correlations. In addition
to these two methods, we provide the results of a third method - SparCC (Sparse Correlations for Compositional Data)(Friedman & Alm 2012), which
is also a method for inferring correlations from compositional data. SparCC estimates the linear Pearson correlations between
the log-transformed components.
References:
Kurtz ZD, Müller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput Biol. 2015 May 7;11(5):e1004226. doi: 10.1371/journal.pcbi.1004226. PMID: 25950956; PMCID: PMC4423992.













{kind=link}
{kind=link}
{kind=link}