| |
FOMC 16S rRNA Taxonomy Assignment Algorithm GitHub Repository
|
| 1. |
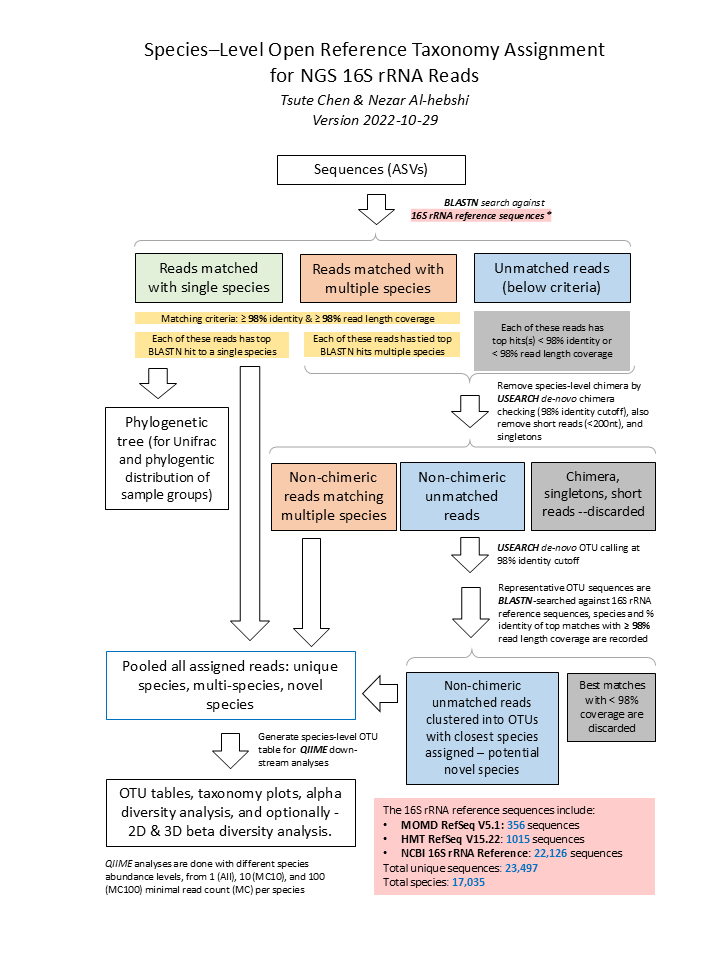
Raw sequences reads in FASTA format were BLASTN-searched against a combined set of 16S rRNA reference sequences - the FOMC 16S rRNA Reference Sequences version 20221029 (https://microbiome.forsyth.org/ftp/refseq/). This set consists of the HOMD (version 15.22 http://www.homd.org/index.php?name=seqDownload&file&type=R ), Mouse Oral Microbiome Database (MOMD version 5.1 https://momd.org/ftp/16S_rRNA_refseq/MOMD_16S_rRNA_RefSeq/V5.1/), and the NCBI 16S rRNA reference sequence set (https://ftp.ncbi.nlm.nih.gov/blast/db/16S_ribosomal_RNA.tar.gz). These sequences were screened and combined to remove short sequences (<1000nt), chimera, duplicated and sub-sequences, as well as sequences with poor taxonomy annotation (e.g., without species information). This process resulted in 1,015 full-length 16S rRNA sequences from HOMD V15.22, 356 from MOMD V5.1, and 22,126 from NCBI, a total of 23,497 sequences. Altogether these sequence represent a total of 17,035 oral and non-oral microbial species.
|
| | |
| |
The NCBI BLASTN version 2.7.1+ (Zhang et al, 2000) [3] was used with the default parameters. Reads with ≥ 98% sequence identity to the matched reference and ≥ 90% alignment length (i.e., ≥ 90% of the read length that was aligned to the reference and was used to calculate the sequence percent identity) were classified based on the taxonomy of the reference sequence with highest sequence identity. If a read matched with reference sequences representing more than one species with equal percent identity and alignment length, it was subject to chimera checking with USEARCH program version v8.1.1861 (Edgar 2010). Non-chimeric reads with multi-species best hits were considered valid and were assigned with a unique species notation (e.g., spp) denoting unresolvable multiple species.
|
| | |
| 2. |
Unassigned reads (i.e., reads with < 98% identity or < 90% alignment length) were pooled together and reads < 200 bases were removed. The remaining reads were subject to the de novo operational taxonomy unit (OTU) calling and chimera checking using the USEARCH program version v8.1.1861 (Edgar 2010)[4]. The de novo OTU calling and chimera checking was done using 98% as the sequence identity cutoff, i.e., the species-level OTU. The output of this step produced species-level de novo clustered OTUs with 98% identity. Representative reads from each of the OTUs/species were then BLASTN-searched against the same reference sequence set again to determine the closest species for these potential novel species. These potential novel species were pooled together with the reads that were signed to specie-level in the previous step, for down-stream analyses.
|
|